
Advancing Comparative Genomics for Human Health: Fauna Bio Collaborates on study to compare 240 mammal genomes
WE ARE EXCITED TO ANNOUNCE THE PUBLICATION OF A NEW STUDY IN NATURE COMPARING THE GENOMES OF 240 MAMMAL SPECIES, INCLUDING 131 NEWLY SEQUENCED SPECIES - THE LARGEST MAMMAL COMPARATIVE GENOMICS PROJECT EVER UNDERTAKEN. FAUNA BIO IS PART OF A MULTI-INSTITUTE COLLABORATION CALLED THE ZOONOMIA CONSORTIUM, WHICH PERFORMED THE WORK. THE STUDY WAS LED BY THE BROAD INSTITUTE OF MIT AND HARVARD AND UPPSALA UNIVERSITY, SWEDEN. IN ADDITION TO BENEFITING BASIC EVOLUTIONARY AND MOLECULAR BIOLOGY RESEARCH, FAUNA BIO IS USING THIS UNPRECEDENTED MULTI-SPECIES RESOURCE TO ADVANCE HUMAN DISEASE BIOLOGY AND TO HELP DISCOVER NEW THERAPEUTICS.

Zoonomia mammal phylogeny. Adapted from figure by Jeremy Johnson, Broad Institute.
THE POWER AND PROMISE OF COMPARATIVE GENOMICS
Even the most diverged organisms share DNA - for instance, humans share many genes with mice, flies, and even yeast. Humans famously share 99% of their genome with chimpanzees, and the DNA that makes up the remaining 1% can help us understand what specifically makes us human. Comparing entire genomes of different organisms not only tells scientists about their shared evolutionary histories, but it can also identify shared characteristics, specific biological differences, and the functions of individual genes or genomic regions.
The more genomes we have to compare, the more accurate and powerful this technique, called comparative genomics, becomes. Genomes are constantly mutating, and while most mutations do nothing, some cause disease and eventually fall out of the gene pool. Comparative genomics relies on the idea of conservation - if a genetic sequence is conserved (remains unchanged), especially over 100 million years of mammal evolution, it’s probably doing something important. While we’ve come a long way from the mid-2000s, when just a handful of mammalian genomes were available, even these early studies were extremely valuable. They allowed us to trim the list of protein-coding genes from 25 thousand down to around 20 thousand, to identify highly conserved non-coding sequence, putting to rest the popular notion of ‘junk DNA’, and to identify hundreds of ‘ultra-conserved regions’, which lack even a single DNA “letter” or base change between humans, rodents, and dogs.

The honey badger. The honey badger genome has been sequenced, but honey badger don’t care.
Compared to those earlier studies, this new dataset is unprecedented in its size thanks to the Zoonomia Consortium’s sequencing of 131 new species. Many of these novel genomes come from some particularly fascinating creatures. My personal favorites include the internet-famous honey badger, which can withstand an onslaught of bee stings and cobra bites, and the grasshopper mouse, which metabolizes scorpion venom into a pain killer. Importantly, this collaboration also documented the genomes of 7 critically-endangered species including Mexican howler monkey, the northern white rhino, and the hirola, an elegant-looking African antelope.
“One of the most exciting features of the Zoonomia Project is that our findings will benefit several different fields. By studying genomes of diverse mammalian species, we are simultaneously powering work to identify the genomic basis of human health and disease and identifying non-human species whose reduced genetic diversity indicates that they should be prioritized for protection," said first author Diane Genereux, a research scientist in the Vertebrate Genomics Group at the Broad Institute.
Comparing the genomes of diverse mammals gives us the ability to observe long-term patterns of mutations, which can identify the DNA bases that are critical for survival. Because people are genetically very similar to each other, even large human datasets with tens of thousands of people cannot tell us which individual bases are functional - the vast majority of the genome would lack mutations and look identical between every person in the study. This means that we would have a very limited understanding of which bases in the human genome are functional using human data alone. But by comparing 240 mammals, we can expect that almost every DNA base should be mutated in at least one species by chance and those bases that are identical are very likely to be important for survival. These same bases have a high likelihood of contributing to disease in people if they are mutated, so identifying them is extremely valuable to medical genetics.
COMPARATIVE GENOMICS AND HUMAN DISEASE
Comparative genomics can also help us understand the genetic basis of human disease. In 2011, a study of 29 mammals identified overlap between conserved regions and mutations implicated in human disease. Indeed, a 2015 study showed that conservation in mammals is currently our best predictor of whether or not a DNA sequence contributes to disease heritability.
Looking forward, our understanding of whether single DNA bases are conserved will be critical for human medical genetics and biotechnology. A senior author on the study, Kerstin Lindblad-Toh of Uppsala University, SciLifeLab and the Broad Institute said that “the comparison of the genomes from the 240 mammals will help geneticists to identify the mutations that lead to human diseases”. Currently, to learn about heritable diseases, like diabetes, scientists compare hundreds of thousands of human genomes from people with and without the disease, implicating regions of the genome that are likely involved. However, these regions are often quite large, containing many genes, and identifying the responsible gene or mutation in a region is still more of an art than a science. Knowing which single DNA bases are conserved across mammalian evolution will help us pinpoint specific disease-causing mutations and genes and eventually identify new therapeutic targets for drug design.
COMPARING GENOMES TO FIND THE BASIS FOR SHARED TRAITS AND ADAPTATIONS
Beyond pinpointing disease-promoting mutations, the Zoonomia data will also lead to a better understanding of gene function. It surprises many people - even some biologists - to learn that we have little idea what 20% of protein-coding genes actually do. Traditionally, scientists investigate gene function by breaking the gene in a model organism (generally a mouse), and then determining what’s different about that mouse. However, this approach doesn’t work easily for all genes or for all traits, especially for traits that do not exist in model organisms. The new Zoonomia dataset affords us the opportunity to take a highly-powered alternate approach. We can now ask: “what is similar about the genomes of mammals with a given trait?”
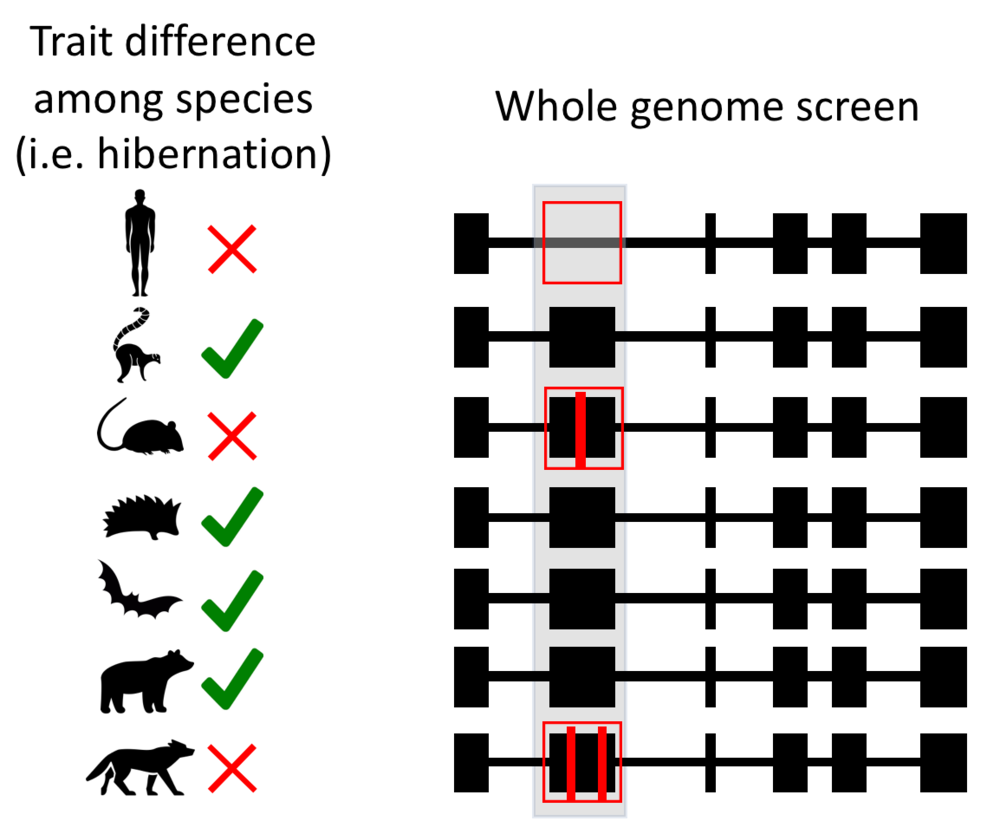
Forward genomics. A method for matching species traits to genes. Figure adapted from https://www.mpi-cbg.de/research-groups/current-groups/michael-hiller/research-focus/
A method called forward genomics, pioneered by Michael Hiller, a Fauna Bio collaborator, and Gill Bejerano, allows researchers to identify the genes that govern key traits that differ among mammals. Given that a trait is ancestral to mammals, and some mammals have lost the trait, important genes will be highly conserved in species that retain the trait, but will be broken or mutated in those that have lost it. For example, Hiller and Bejerano used the method to identify a gene required for vitamin C synthesis. The gene, called Gulo, is broken in humans and all other mammals whose bodies can’t synthesize the vitamin, but is preserved in animals who can.
"Zoonomia is designed to find the parts of our DNA that haven't changed over a hundred million years of evolution, and the parts that are changing in interesting ways. How are aquatic mammals, or hibernating mammals, or flying mammals, genetically different from humans, and can that give us a clue about how they survive in environments that would be dangerous, and even fatal, for humans? With the Zoonomia alignment, we can start to figure that out," said co-senior author Elinor Karlsson, director of the Vertebrate Genomics Group at the Broad Institute of MIT and Harvard and professor at the University of Massachusetts Medical School.
IDENTIFYING CONSERVED GENES RESPONSIBLE FOR HIBERNATION
Applying hibernation biology to human therapeutics is one of Fauna Bio’s primary R&D goals. Now, in collaboration with researchers from the Broad Institute, The University of Nevada and The Center for Translational Biodiversity Genomics Frankfurt, we are using the Zoonomia dataset and the principle of forward genomics to better understand and someday harness mammalian hibernation biology. While the concept may seem counterintuitive, there is extensive evidence that hibernation is actually an ancestral trait in mammals. Hibernators can be found in every major family of mammals, including primates. This suggests that the trait was lost somewhere in the primate lineage prior to the divergence of humans. There is also new fossil evidence that an early mammal-like reptile called Lystrosaurus was a hibernator.
Given that hibernation is ancestral, we can find genes and genetic pathways that are important for hibernation by finding genes that are especially conserved (i.e. have changed the least) in animals that hibernate. The Zoonomia dataset contains at least 36 animals with clear evidence of hibernation (it’s still not known whether or not some animals hibernate). We expect to uncover not only genes necessary to induce and maintain the hypometabolic state of hibernation, but also genes needed to protect animals from the many hazards associated with hibernating: muscle/bone atrophy from lack of use, extreme obesity, and heart damage from dramatic changes in blood flow to name just a few. Fauna Bio is currently focused on the protective adaptations of hibernation to identify drug targets for humans, but this initial study just scratches the surface of traits that could be investigated with this dataset, and we are excited about the many more that are sure to follow.